In Time to Help Jessie
By Quinn Eastman

The Stinchcomb children, Jessie, Jenna, Jordan, and Jake, with parents Pam and Tony. Family photo
For most of her life, 19-year-old Jordan Stinchcomb’s parents thought she had cerebral palsy.
Jordan was born by emergency Cesarean section with the umbilical cord wrapped around her neck, and had to be resuscitated. Her parents assumed that lack of blood flow and oxygen at birth had damaged her brain, causing her inability to walk and speak.
Pam and Tony Stinchcomb, who live in Hoschton, Georgia, had two more children, Jenna and Jake, who were born without similar difficulties. Then when Jordan was 13, her youngest sister Jessie was born, after an almost perfect pregnancy.
“We knew something was not right,” says Pam, a physical therapist. “She cried for the first six months and would scream if we put her on her stomach.”
Jessie missed early milestones such as being able to roll over or sit up by herself. At around six months of age, she started wheezing because of an airway restriction. Her parents thought they saw hints that Jessie was experiencing seizures, which Jordan had started having at age 11. They decided to seek help and an explanation.
Neurologists they consulted suspected Jessie had a congenital condition: perhaps Rett syndrome, a neurodevelopmental disorder that mainly affects girls, or a mitochondrial disorder. A muscle biopsy and other invasive procedures were performed. Months went by, while tests for known diseases came back negative.
 |
|
When the Stinchcombs were referred to Emory, they encountered a delay when a respected geneticist who specialized in rare disorders, Paul Fernhoff, associate professor of human genetics and pediatrics and the medical director of human genetics at Emory University Hospital, died unexpectedly.
When medical geneticist Michael Gambello arrived at Emory from Houston in 2012, he recommended that the family proceed with whole exome sequencing for Jessie. It wasn’t a difficult decision. At that time, whole exome sequencing was emerging as a powerful technique for finding the mutations that might be responsible for a disease that looks like an inherited condition, when tests for known genetic disorders have not proven helpful.
Several weeks later, the Emory Genetics Laboratory, under the direction of Madhuri Hegde, found something, but it needed additional confirmation. Gambello asked whether Jordan’s DNA could also be tested.
Pam and Tony were surprised, since they had thought Jordan’s condition was caused by the circumstances of her birth.
“It made sense 19 years ago,” Pam says. “Until Jessie was born, we never had a need to look for anything else.”
Gambello told the Stinchcombs about the sequencing results, but he was able to tell them little about the disease or Jessie’s prognosis.
Pam Stinchcomb recalls the conversation going something like this:
“We’ve learned that your daughter has N-glycanase-1 deficiency. It’s only been identified recently.”
“What is that?”
“We really don’t know. There is another patient reported in the medical literature, who has symptoms almost identical to Jessie.”
The disorder was not something doctors were familiar with, even geneticists who deal with rare diseases.
“At that point, there was one publication out there describing a single patient with NGLY1 deficiency,” Gambello says. “It could have been wrong. But we had two affected siblings, and the parents were carriers.”
N-glycanase 1 is an enzyme—part of a complex apparatus that processes proteins— that cell biologists have studied for years in the laboratory. However, it was highly unusual for doctors to see not one, but two living patients in the same family who appeared to lack this enzyme.
Coincidentally, shortly after talking with the Stinchcombs, Gambello received an email from a genetic counselor he had worked with in Houston, seeking advice for another family with a child with NGLY1 deficiency.
From this communication, he and the Stinchcombs learned about several other children around the world with the same genetic deficiency, whose cases had not yet been published in medical journals.
Several of the affected families share the same exact genetic mutation in the NGLY1 genes, even though the families do not appear to be related, beyond sharing European ancestry. This may indicate a “founder effect” reaching back many generations.
Having felt isolated and alone, the families were hungry for information from the medical community and from each other.
It’s hard enough for your child to have a chronic or debilitating condition, let alone one that doctors are stumped by.
One of the fathers, Matt Might, a computer scientist at the University of Utah, wanted to find other families dealing with the same challenges.
After an odyssey of diagnostic missteps led to the identification of NGLY1 deficiency in his son, Bertrand, Might wrote the essay “Hunting down my son’s killer” on his personal website.
“Aside from severe jaundice, Bertrand was normal at birth,” Might writes. “For two months, he developed normally. At three months, his development slowed, but it was ‘within normal variations.’ By six months, he had little to no motor control. He seemed, as we described it, ‘jiggly.’ Something was wrong.”
With his post amplified by social media sites like Reddit, over the next year the Mights found nine other families with a child with NGLY1 deficiency.
A Lack of Tears
The Mights’ quest was highlighted by science writer Seth Mnookin in the New Yorker (“One of a Kind”) and by CNN (“Kids who Don’t Cry”).
“For families in this situation, the first thing a diagnosis does is to stop their frustrated progress from doctor to doctor,” says Stephen Warren, chair of human genetics at Emory. “And it’s a starting point for figuring out the rest, even if that may take time. The parents can organize and compare experiences.”
About a year after Jessie and Jordan were diagnosed, the Stinchcombs met the Mights, the Wilseys, and other affected families at a 2014 conference devoted to NGLY1 in San Diego at Sanford Burnham Prebys Medical Discovery Institute.
Pam Stinchcomb says she now regularly compares notes with the other parents on caring for their children. “Just having that support is wonderful,” she says. “It’s like having a little NGLY1 family.”
The group has continued to grow. As of the summer of 2015, geneticists have been able to confirm 27 cases of NGLY1 deficiency by sequencing. Lynne Wolfe, coordinator of a study at the National Institutes of Health, and her team have thoroughly examined 12 of those individuals, including both Jessie and Jordan Stinchcomb.
Patients with NGLY1 deficiency appear to have several core symptoms: involuntary movements, hypotonia (low muscle tone), and an inability to produce tears. EEG abnormalities are common in many of the children identified so far, with overt seizures present in about half.
Most of the children have some level of developmental delay and problems with auditory processing or expressive language, Wolfe says. Still, the degree of developmental delay is variable. Some affected children have a limited vocabulary. Jessie can communicate through whines and glances, but does not speak recognizable words.
Liver disease was thought to be a common feature, but in several children, liver abnormalities have faded as they have become older, Wolfe says.
For the Stinchcombs, seizures have been a dominant and troubling feature of the disorder for Jordan, at least in the past few years.
Visiting Jessie and Jordan
When entering the Stinchcombs’ home, visitors may notice a chirping, bird-like sound.
“Oh, that’s just Jessie, watching her favorite DVD,” Tony Stinchcomb says.
Jessie enjoys listening to “Clifford the Big Red Dog.” She seems to depend on it. When her father brings her into the family’s kitchen to eat some applesauce, she does fine as long as the DVD is playing on a rubber-cushioned iPad. She squeals with delight while sitting on Tony’s lap and playing with him. But when he turns Clifford off for just a moment, she becomes visibly distressed.
When seated, Jessie’s arms are in constant motion. Her room, which she shares with Jordan, is full of large pillows and decorated with Frozen movie posters. She can push herself up in her crib and move around by rolling on the pillows but can’t crawl in a sustained way.
When Jessie does push herself up, her weight rests on the sides of her feet in what looks like an awkward ankle twist. She has corrective braces but finds them painful, her parents say. As part of doctors’ efforts to help her stand, she has received botulinum toxin injections in her legs.
The edges of Jessie’s and Jordan’s eyes often look red. They receive eye drops several times per day to compensate for the lack of tear production.
Jessie and Jordan are both on a specialized formula diet. They have chronic gastrointestinal problems: constipation and reflux. Tony says that he sometimes needs to change his clothes four times per day because of spit-up.
Even before her sister was born, Jordan had taken an anti-seizure medication, levetiracetam, with doctors occasionally increasing the dose. Her parents became concerned that she seemed to be falling asleep repeatedly and had her taken to a hospital to perform a 72-hour EEG, in which “abnormal brain activity” was detected. In February 2014, Jordan had a frightening series of seizures in which she stopped breathing.
“Her lips were turning scary blue,” recalls Tony, who yelled for his son, Jake, to call 911 while he tried to revive her. Jordan was unable to travel to California that spring to meet other NGLY1-affected families.
Then in March of 2015, she experienced more than a dozen seizures. For the several days that month she spent in the hospital, Jordan was non-responsive and had no muscle tone, her mother recalls. “She did wake up from time to time, look at us, and even smile,” Pam says.
Doctors managed to find a drug, phenytoin, that could stop the cluster episodes, and Jordan is back at home, taking that drug and other anti-seizure medications regularly.
Before the seizures
Jordan is now less mobile than Jessie, although that wasn’t always the case. She used to be able to stand by herself and move around with a walker. Before her recent seizure episodes, Jordan could recognize her printed name and could roll a ball back and forth or engage in tug-of-war play with her sister, her parents say. She enjoyed going to her brother Jake’s weekend baseball or basketball games, where she would devour hot dogs, and she liked to splash around in the family’s pool.
When Jordan was a small child, her parents took her several times to New Orleans for hyperbaric oxygen treatments. They say the oxygen treatment helped her become more alert, sleep through the night, and track objects with her eyes more accurately, although the general effectiveness of this type of treatment has been debated.
Jordan attended a specialized day care center in nearby Gainesville and then public schools. Her parents pulled her out of public school after she experienced several falls and had frequent respiratory infections. For a while, her parents worked grueling split shifts to take care of her. Then Tony decided to stop working at a nearby rock crushing plant, where his relatives still work, and care for Jordan full-time.
In May 2014, after the Stinchcombs returned from meeting other NGLY1-affected families in California, a community fundraiser enabled them to buy a hand-icapped-accessible van that can transport both girls’ wheelchairs.
Around the same time, a friend’s foundation built the Stinchcombs a spacious shower, which allows them to bathe Jordan more easily.
Chain of sugars
The NGLY1-affected families have sparked a surge of research into how the absence of the N-glycanase 1 enzyme affects cells and the entire body, with an eye toward possible treatments.
The enzyme encoded by the NGLY1 gene is thought to be involved in protein quality control. Some form of this enzyme can be found in yeast, worms, flies, and even plants. NGLY1’s basic function is to remove a chain of sugars from unfolded proteins. If proteins become unfolded while they are being synthesized, the cell destroys them in the proteasome, a kind of molecular “garbage disposal.”
 |
|
|
Medical geneticist Michael Gambello recommended that the family proceed with whole exome sequencing for Jessie, since he suspected a genetic disorder. Her older sister, Jordan, was discovered to have the disorder as well. Photo by Jack Kearse. |
Up until the discovery of the NGLY1-affected families, scientists who studied the enzyme thought that the chain of sugars acted like a trash tag, telling the cell what to do with some of the unfolded proteins. If the NGLY1 enzyme is absent from cells, then the garbage disposal might go astray. But which other proteins’ production or processing is disturbed and what causes the array of symptoms, nobody knows.
Because nervous system development is derailed, it’s a good bet that NGLY1-dependent changes can be seen in neurons and/or glial cells, Emory’s Michael Gambello says.
He has been working on engineering mice that are deficient in NGLY1. Gambello’s preliminary findings show some differences between human patients and the mice, in that the mice don’t seem to have a movement disorder.
Still, it is common for geneticists to find discrepancies between human genetic disorders and their mouse models. Gambello is also checking whether the NGLY1-deficient mice are more prone to seizures.
Hudson Freeze and his colleagues at Sanford Burnham Prebys Medical Discovery Institute have been examining cells obtained from NGLY1-affected patients. Their findings are going against previous assumptions about how the enzyme deficiency would affect cells, he says.
His team has looked for signs that unfolded proteins accumulate in patient cells and has not seen them. The same goes for accumulation of cellular structures (lysosomes) where proteins are recycled, he says.
These findings illustrate that scientists are still figuring out how NGLY1 is involved in protein processing and trash removal.
Several other rare diseases are caused by inherited problems with enzymes whose job it is to add chains of sugars to proteins. NGLY1 does the opposite and removes the sugar chains.
Despite the divergence, these deficiencies both interfere with brain development, and affected individuals often have developmental delays, movement disorders, and seizures.
Fathers Matt Might and Matt Wilsey have written that the NGLY1 disorder’s rapid emergence is an example of how discovery can be sped up when families, physicians, and geneticists work together to determine causes and potential treatments.
With the families comparing notes, ideas are exchanged, ranging from the use of cocoa extract to carnitine, a dietary supplement that the Stinchcombs have found helps with Jessie’s gastrointestinal problems.
With the choice of anti-seizure drugs, an issue that was urgent for the Stinchcombs, it’s less clear that crowd-sourcing has made a difference.
“It’s hard to say what ‘usually works’ when the number of affected patients is so small,” Gambello says. “Even with other types of epilepsy, the process of choosing effective anti-seizure medication is often one of trial and error, and this is a new disorder.”
 |
Freeze’s team has already tested one option for treatment of NGLY1 deficiency, without success.
Many inherited diseases, this one included, involve introduction of a premature stop signal into the part of a gene that encodes a protein. Read-through agents, a group of compounds that includes some antibiotics and a drug in cystic fibrosis clinical trials, are supposed to coax the cell into “reading through” the genetic glitch. However, Freeze found that these compounds didn’t restore enzyme activity in NGLY1-deficient human cells.
Other potential drug treatments have been proposed, such as EPI-743, a potent antioxidant developed for inherited mitochondrial diseases.
Freeze has been testing this drug on NGLY1-deficient patient cells as well. Japanese researchers led by Tadashi Suzuki recently found that inhibiting another enzyme (ENGase) could compensate for the absence of N-glycanase 1 in mouse cells.
Additional insights into the variability of NGLY1 deficiency may emerge from the NIH study, with Jordan being one of the oldest known affected individuals at age 19.
“Jordan is writing the book on this disease,” Pam Stinchcomb says.
The idea that NGLY1 deficiency may be a progressive disorder that worsens with time deeply troubles the Stinchcombs, who were high school sweethearts before marrying and starting a family.
Pam regrets the losses their oldest daughter has endured already, and says, “We don’t accept that it just progresses.”
Still, when asked whether it was worth going through the process of diagnosis, Pam says, “Absolutely. I wouldn’t change anything about that. We know something about what we’re dealing with, and we’re not alone. Even if there is no treatment now, I continue to pray that we will get there while Jessie will be able to benefit.”
EMORY GENETICS LABORATORY
Finding the genes associated with rare disorders
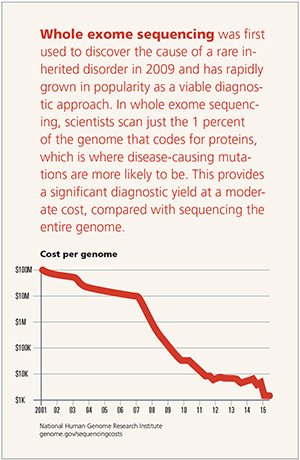
While it is only a small part of the genome, the exome accounts for 85 percent of disease-causing gene alterations.
With nearly five decades of clinical experience, Emory Genetics Laboratory (EGL) is the oldest clinical genetics lab in the U.S. and was one of the first labs in the world to launch whole exome sequencing in 2011.
Since then, exome sequencing has proven to be an invaluable diagnostic technique for clinicians seeking molecular confirmation of a suspected genetic disorder.
“We are quickly moving away from the idea of single genes causing single disorders. One gene can cause many disorders, and many genes can cause one disorder,” says EGL executive director and clinical molecular geneticist Madhuri Hegde. “We have had to change our ideas about data analysis and interpretation of the data.”
The EGL is part of Emory’s Department of Human Genetics, which is home to the National Down Syndrome project and serves as one of three national fragile X research centers. EGL contributes to the “Free the Data” movement by giving the clinical and research community open access to its online variant (mutation) database, EmVClass. EGL also serves as the follow-up laboratory for the Georgia Newborn Screening Program.
 |
|
|
EGL Executive Director and clinical molecular geneticist Madhuri Hegde. |
Hegde has a special interest in the development of next-level sequencing strategies for rare disorders—her clinical work focuses on identifying new genes for muscular dystrophy and developing a follow-up program for newborn confirmatory testing for Duchenne muscular dystrophy.
Lora Bean, lab director at EGL, and Lindsey Mighion, genetic counselor at EGL, remember the Stinchcomb sisters well, and use the case as an example of the diagnostic advantage of whole exome sequencing for patients with suspected genetic disorders.
At first, the lab had a “proband only” sample from a 2-year-old girl with severe developmental delay. In genetics, the proband is the first affected family member who seeks medical attention for a genetic disorder—in this case, Jessie. Through exome sequencing, they found that each of her parents was a carrier, and her older sister, Jordan, also had the changes in the NGLY1 gene.
”At the time, only one paper had reported this gene,” says Mighion.
They reached out to colleagues in other labs and ultimately found eight affected individuals with NGLY1 deficiency.
“This is a case that we highlight for a couple of reasons—it’s why data sharing is so important. Only because of the large collaboration were we able to show that this gene was truly associated with disease and report this,” Mighion says. “Also, it shows the importance of constantly curating, so we can add genes that used to be of unknown significance to our genes-associated-with-disease categories.”
Genomic sequencing will likely become a routine component of one’s medical “family history,” providing the important variants inherited from each parent and the new mutations that contribute to disease susceptibility.
“This allows us to find better, individualized answers related to rare genetic diseases for more patients with complicated medical issues than we ever could before,” says Dawn Laney, EGL genetic counselor and director of the Emory Genetic Clinical Trial Center.—Mary Loftus
For more: geneticslab.emory.edu.





